

HEMATOLOGIA

DIAGNÓSTICO1
O diagnóstico de hemofilia deve ser pensado sempre que há histórico de sangramento fácil, após pequenos traumas, ou espontâneo.1
Os sintomas podem se manifestar em hematomas subcutâneos nos primeiros anos de vida ou sangramentos musculares e/ou articulares em meninos acima de dois anos. Além disso, observa-se o histórico de sangramento excessivo após procedimentos cirúrgicos ou extração dentária.1
É importante lembrar que, embora o histórico familiar esteja frequentemente presente, em até 30% dos casos pode não haver antecedentes familiares com hemofilia.1
O coagulograma com alargamento do tempo de tromboplastina parcialmente ativada (TTPa) e tempo de protrombina (TP) normal é observado na grande maioria das vezes, com exceção de alguns casos de hemofilia leve, em que o TTPa permanece normal.1
O diagnóstico confirmatório é realizado por meio da dosagem da atividade coagulante do fator VIII (hemofilia A) ou fator IX (hemofilia B).1
Os exames laboratoriais completos, assim como as técnicas empregadas e a avaliação dos resultados, estão disponíveis no Manual de Diagnóstico Laboratorial das Coagulopatias Hereditárias e Plaquetopatias.1
Diagnósticos diferenciais
Dentre os diagnósticos diferenciais devem ser lembradas outras doenças hemorrágicas, como a doença de von Willebrand, considerado um importante diagnóstico diferencial para a hemofilia A.1 A tabela abaixo lista os exames necessários para a investigação inicial das principais coagulopatias.1
Diagnóstico diferencial das coagulopatias
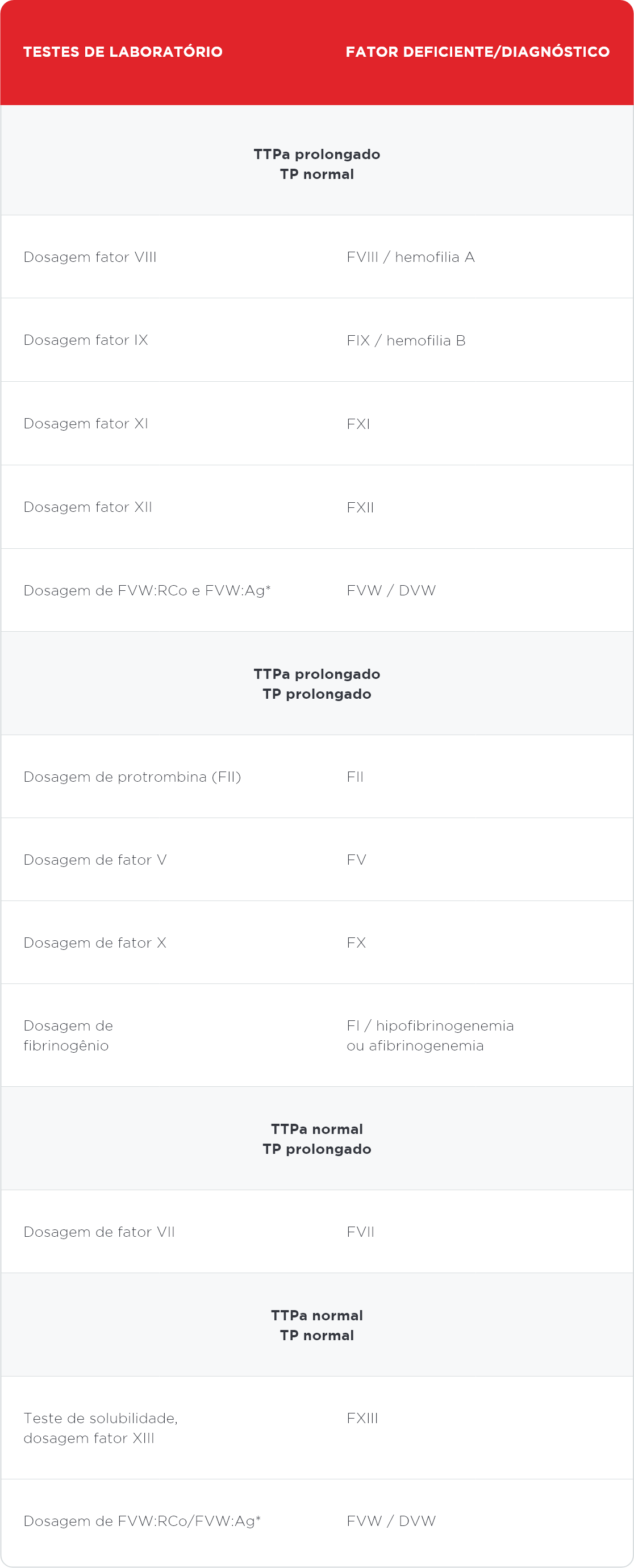
* Outros testes diagnósticos para a doença von Willebrand incluem o tempo de sangramento pelo método de Ivy e a agregação plaquetária com ristocetina (RIPA)
• TTPa, tempo de tromboplastina parcial ativado
• TP, tempo de protrombina
• F, fator
• FVW:RCo, cofator ristocetina
• FVW:Ag, fator (antígeno) de von Willebrand
• FVW, fator von Willebrand
• DVW, doença von Willebrand
É importante lembrar que existem deficiências combinadas de fatores. Entre os casos, pode ocorrer deficiência combinada de fator VIII e fator V (TTPa e TP alargados com a diminuição da atividade de fator VIII e fator V), além da deficiência combinada dos fatores dependentes da vitamina K (fatores II, VII, IX, X, proteína C e proteína S).
CARACTERÍSTICAS
As complicações da hemofilia são caracterizadas pelos tipos de sangramentos: espontâneos, prolongados em ferimentos pequenos e excessivos após procedimentos cirúrgicos.
HEMOFILIA A
A hemofilia tipo A é uma doença hemorrágica hereditária ligada ao cromossomo X, caracterizada pela deficiência ou anormalidade da atividade coagulante do fator VIII.
HEMOFILIA B
A hemofilia tipo A é uma doença hemorrágica hereditária ligada ao cromossomo X, caracterizada pela deficiência ou anormalidade da atividade coagulante do fator VIII.
HEMOFILIA A ADQUIRIDA (AHA)
A hemofilia tipo B é uma doença hemorrágica hereditária ligada ao cromossomo X, caracterizada pela deficiência ou anormalidade da atividade coagulante do fator IX.
DIAGNÓSTICO
O diagnóstico de hemofilia deve ser pensado sempre que há histórico de sangramento fácil, após pequenos traumas, ou espontâneo.
Referências bibliográficas
-
Brasil. Ministério da Saúde. Secretaria de Atenção à Saúde. Departamento de Atenção Especializada e Temática. Manual de hemofilia / Ministério da Saúde, Secretaria de Atenção à Saúde, Departamento de Atenção Especializada e Temática. - ed., 1. reimpr. -Brasília : Ministério da Saúde, 2015.


